1.结构优化(geo_opt)
以MS的forcite模块为例进行结构优化
打开MS并导入cif文件
点击MS菜单选择【Modules】——【forcite】——【calculation】
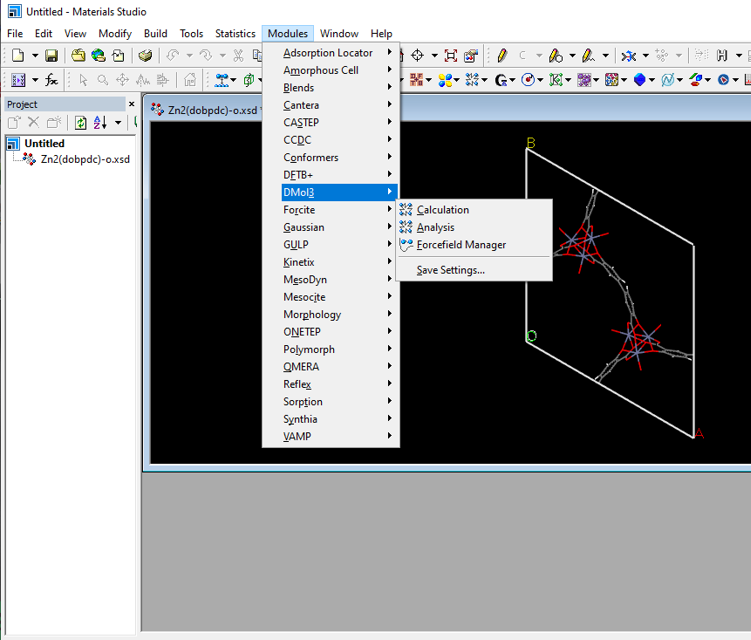 将【task】选项改为Geometry optimization(点击more即可打开左边的窗口)
将【task】选项改为Geometry optimization(点击more即可打开左边的窗口)
在【setup】选择合适的精度(我这里选取Smart, Ultra-fine的精度)
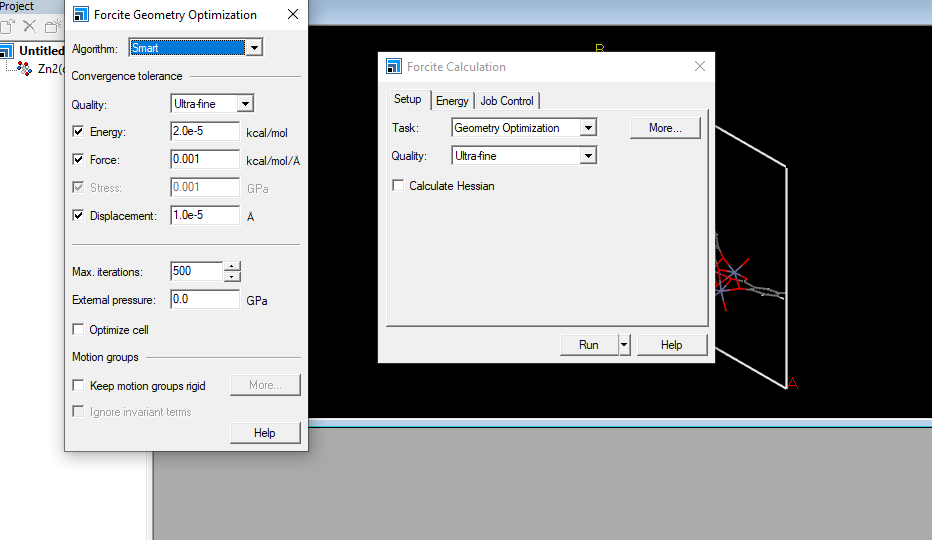
在【energy】选项中,力场(forcefiled)选择universal,质量(quality)选择ultra-fine
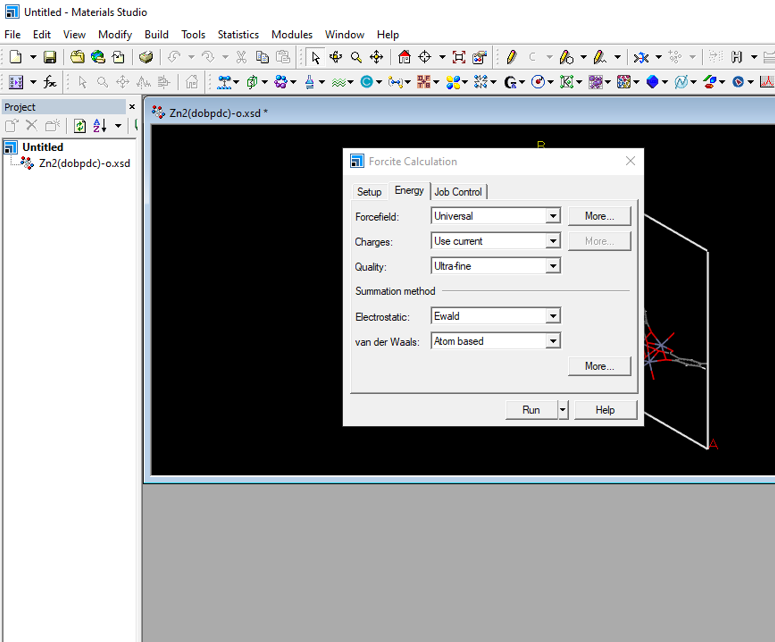
点击【run】
产生结构优化文件
选择文件夹中.xsd后缀的文件
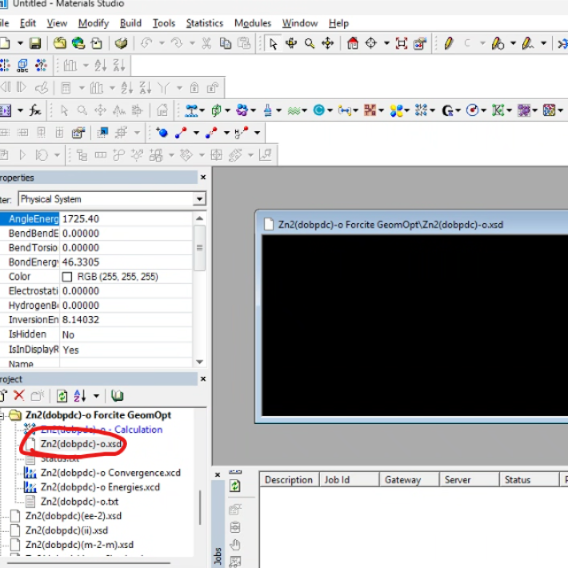
点击【file】——【export】(格式选择为cif格式)
至此,我们获得了结构优化后的cif文件。
2.进行氦气孔隙率模拟
编辑raspa input文件(以下为示例文件)
运行文件相对位置
1
2
3
4
5
6
| RASPA_Simulation/
├── simulation.input
├── force_field_mixing_rules.def
├── helium.def
├── pseudo_atoms.def
├── Zn2(dobpdc)-o.cif
|
simulation.input
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
| SimulationType MonteCarlo
NumberOfCycles 30000
NumberOfInitializationCycles 3000
PrintEvery 1000
RestartFile no
Forcefield local
UseChargesFromCIFFile yes
CutOff 10.5
Framework 0
FrameworkName Zn2(dobpdc)-0 #此处更改为需要进行模拟cif文件名
UnitCells 1 1 4
ExternalTemperature 298.0
Component 0 MoleculeName helium
MoleculeDefinition local
WidomProbability 1.0
CreateNumberOfMolecules 0
|
进行模拟
模拟完成后会产生以下文件,
1
2
3
4
5
6
7
8
9
10
| RASPA_Simulation/
├── simulation.input
├── force_field_mixing_rules.def
├── helium.def
├── pseudo_atoms.def
├── Zn2(dobpdc)-o.cif
├── Movies/
├── Output/
├── Restrat/
└── VTK/ #用来进行可视化的(可有可无)
|
使用9950x的单核模拟,消耗时间约为3mins
打开【output】——【system0】——output文件
查找Average Widom Rosenbluth-weigh
示例:
[helium] Average Widom Rosenbluth-weight: 0.249963 +/- 0.000741 [-]
(0.249963为氦气孔隙率,0.000741为误差)
准备工作完成后,可以进行正式的气体模拟。
3.气体吸附模拟
与氦气孔隙率的模拟类似,只是把氦气替换为你所需要吸附的气体
这里以CO2为例
1
2
3
4
5
6
| RASPA_Simulation/
├── simulation.input
├── force_field_mixing_rules.def
├── CO2.def
├── pseudo_atoms.def
├── Zn2(dobpdc)-o.cif
|
simulation.input文件示例:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
| SimulationType MonteCarlo
NumberOfCycles 1000000
NumberOfInitializationCycles 1000000
PrintEvery 1000
Forcefield local
UseChargesFromCIFFile yes
ChargeMethod Ewald
CutOff 12
Framework 0
FrameworkName Zn2(dobpdc)-o
UnitCells 2 2 4
HeliumVoidFraction 0.249963 #此处填写氦气孔隙率的模拟结果
ExternalTemperature 298
ExternalPressure 100000
ComputeDensityProfile3DVTKGrid yes
WriteDensityProfile3DVTKGridEvery 100000
DensityProfile3DVTKGridPoints 150 150 150
AverageDensityOverUnitCellsVTK yes
DensityAveragingTypeVTK FullBox
Component 0 MoleculeName CO2
MoleculeDefinition local
IdealGasRosenbluthWeight 1.0
TranslationProbability 1.0
RotationProbability 1.0
ReinsertionProbability 1.0
SwapProbability 2.0
CreateNumberOfMolecules 0
|
*raspa网站 (RASPA – iRASPA)
具体参数设置及学习可以参照raspa2的手册(raspa manual)
模拟完成后产生以下文件
1
2
3
4
5
6
7
8
9
10
| RASPA_Simulation/
├── simulation.input
├── CO2.def
├── force_field_mixing_rules.def
├── pseudo_atoms.def
├── Zn2(dobpdc)-o.cif
├── Movies/
├── Output/
├── Restrat/
└── VTK/ #用来进行可视化的(可有可无)
|
使用9950x的单核模拟消耗约24h
之后可以在【output】——【system0】——output文件 寻找感兴趣的结果